주메뉴 바로가기
본문 바로가기
하단 영역 바로가기
사업소개
질병관리청 국립보건연구원
보건복지부
데이터베이스
데이터 현황
데이터 조회
주요서비스
주요서비스
데이터신청절차
수수료 안내
웹분석
현장분석
데이터 분석 예약
알림·활동
공지사항
자료실
연구성과공유
KOR
KOR
ENG
데이터활용
데이터활용
사업소개
주요서비스
데이터베이스
데이터활용
알림·활동
오픈소스
자체개발
오픈소스
데이터 분석도구
오픈소스
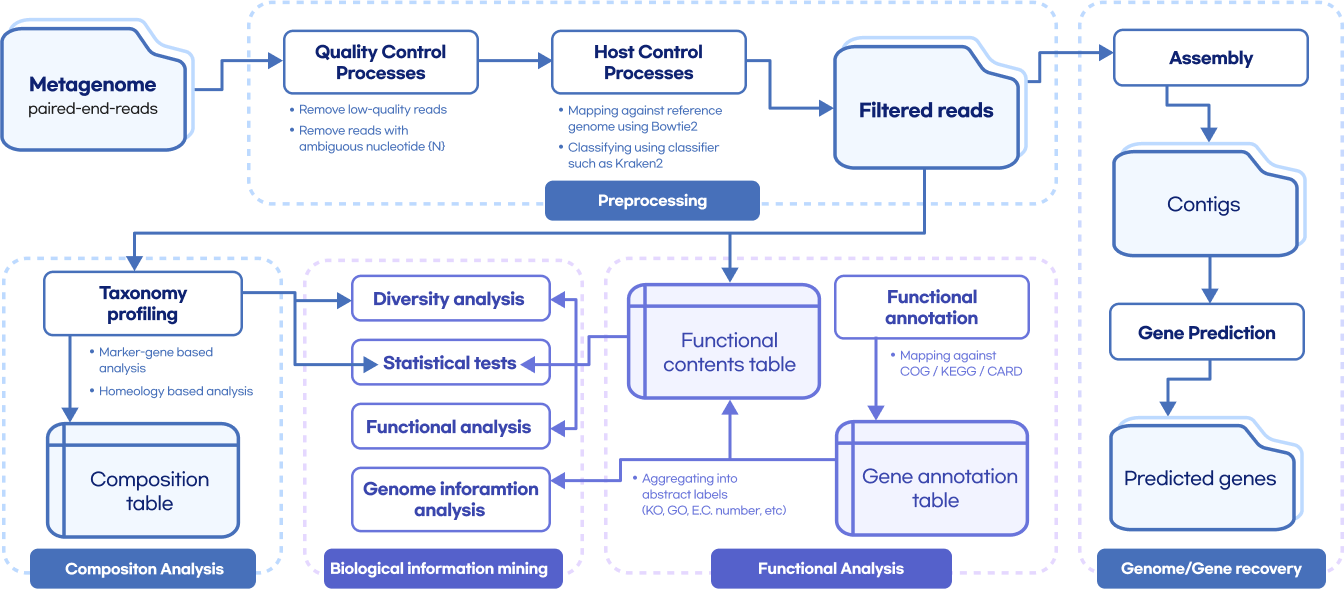
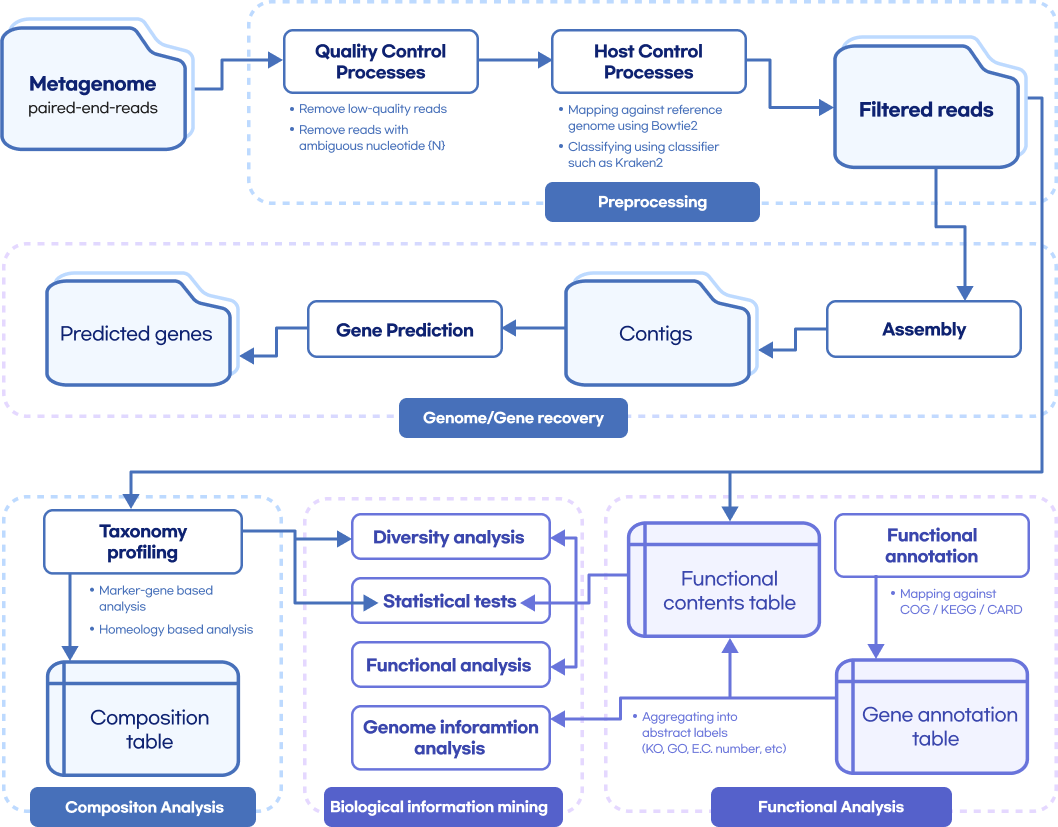
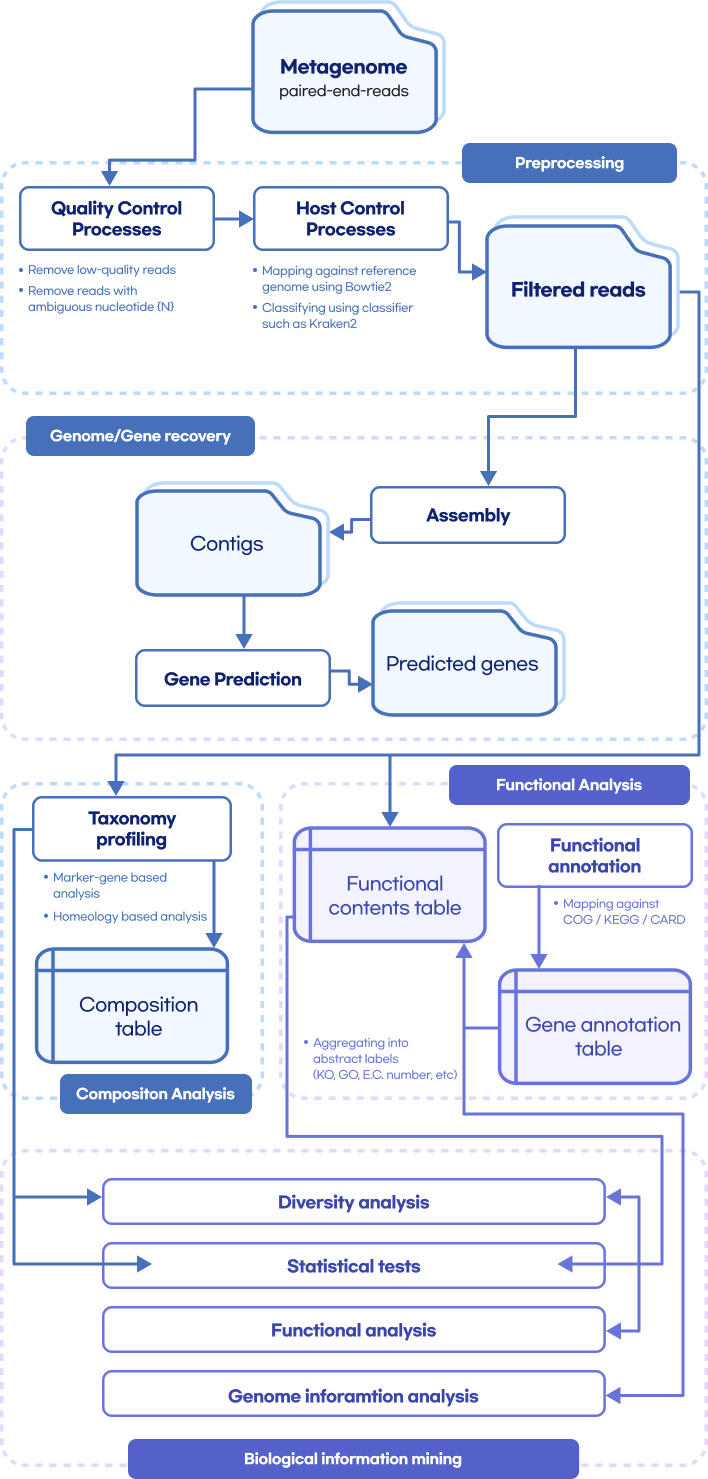
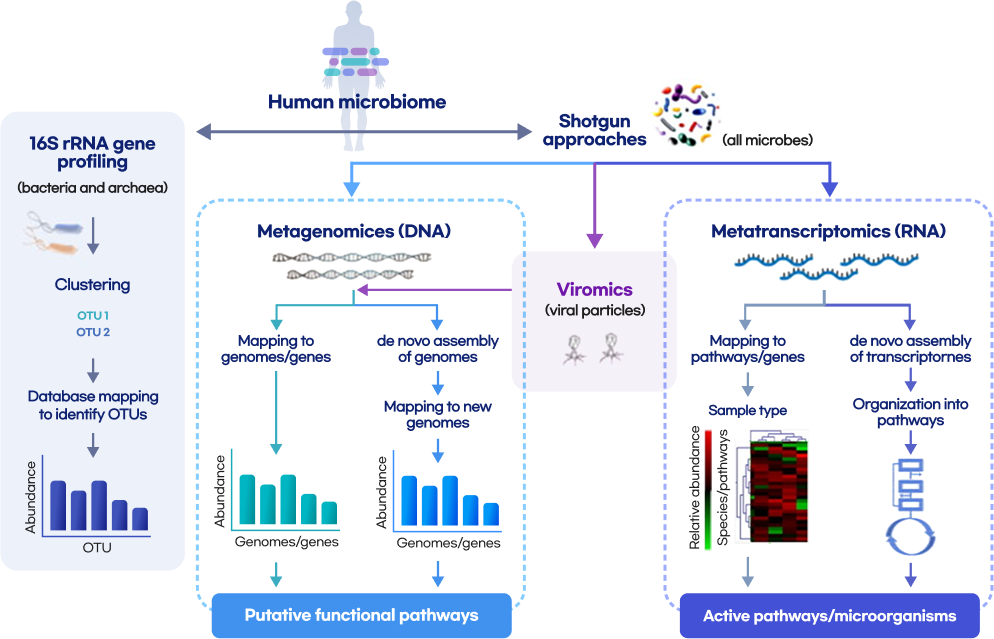
Whole metashotgun sequencing analysis Workflow
Quality Control
(FastQC, Trimmomatic, TagCleaner)
Remove host contamination
(bowtie2, samtools, bedtools)
Taxonomic Classification
(kraken, kaiju, mOTU, BLAST)
Genome Assembly
(MEGAHIT, metaSPAdes, MetaVelvet)
Contig binning
(CONCOCT, MetaBAT2, MaxBin2)
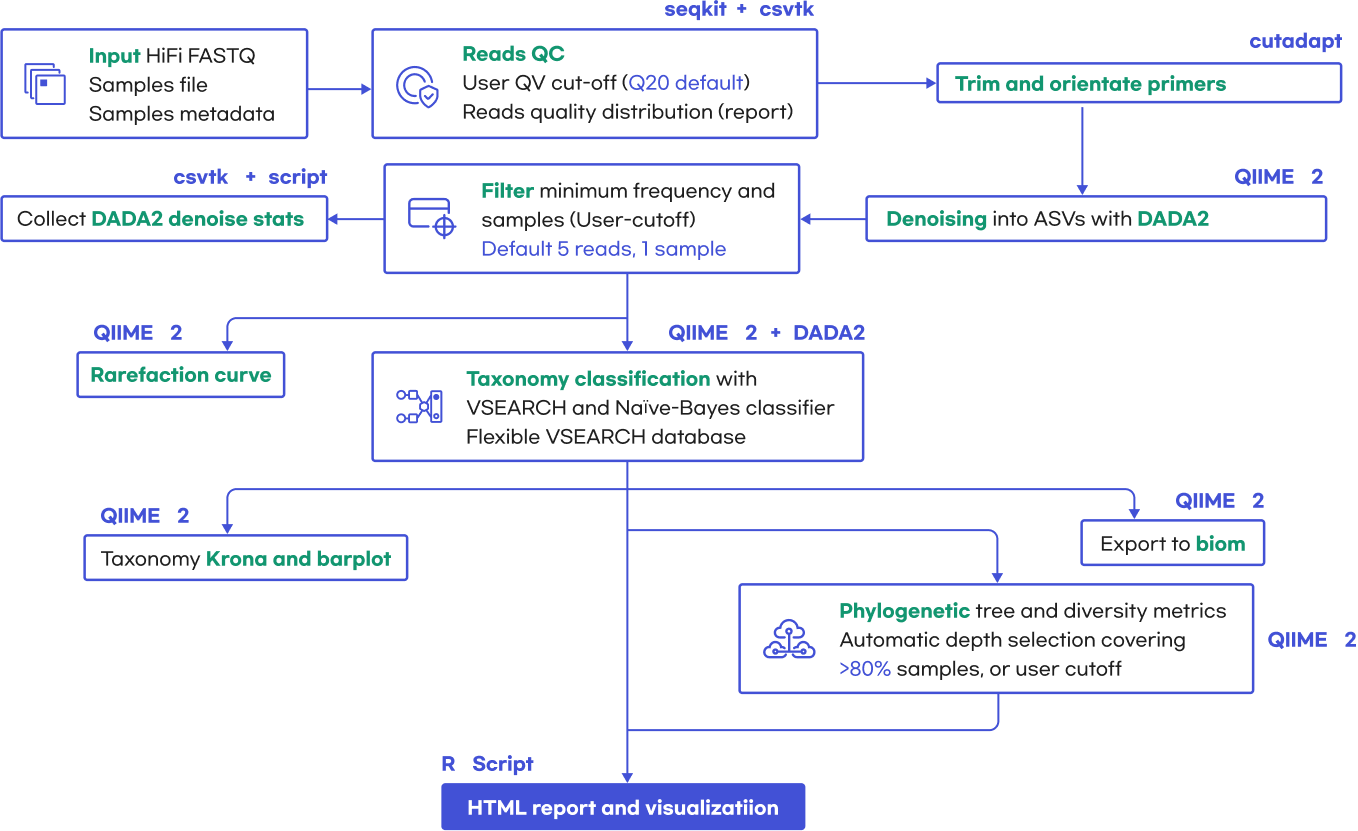
16S Sequencing Analysis
Quality control (QC)
reads > 20,000(분변검체), 5,000(그 외 검체)
ampliicon size(bp) > 1,200(Long read), 400(Short read)
16S amplicon sequencing analysis Workflow : QIIME2를 사용하여 FASTQ file을 분석
Demultiplex samples
Sequence Quality check with FastQC
Decontaminate and Trim sequences and cut adapters, primers, etc
ASV Assignment with DADA2
Filter and Trim
Merge Forward + Reverse Reads
Create Sequence Table and Remove Chimeras
Remove Chimeras
Assign Taxonomy to ASVs (Database: NCBI, SILVA, Greengenes를 권장)
Save DADA2 Output for future analysis
Statistical Analysis using R programs, etc.
오픈소스 분석
시퀀싱 플랫폼
Illumina Miseq/Novaseq
(Short read)
Pacbio Sequel2
(Long read)
×
사업소개
질병관리청 국립보건연구원
보건복지부
데이터베이스
데이터 현황
데이터 조회
주요서비스
데이터신청절차
수수료 안내
웹분석
현장분석
데이터 분석 예약
알림·활동
공지사항
자료실
연구성과공유
×
사업소개
질병관리청 국립보건연구원
보건복지부
데이터베이스
데이터 현황
데이터 조회
주요서비스
데이터신청절차
수수료 안내
웹분석
현장분석
데이터 분석 예약
알림·활동
공지사항
자료실
연구성과공유